UNSATURATED OR UNSATURATED HYDROCARBONS OF THE ETHYLENE SERIES
(ALKENES OR OLEFINS)
Alkenes, or olefins(from Latin olefiant - oil - an old name, but widely used in chemical literature. The reason for this name was ethylene chloride obtained in XVIII century, - liquid oily substance.) - aliphatic unsaturated hydrocarbons, in the molecules of which there is one double bond between the carbon atoms.
Alkenes contain fewer hydrogen atoms in their molecule than their corresponding alkanes (with the same number of carbon atoms), therefore such hydrocarbons are called unlimited or unsaturated.
Alkenes form a homologous series with the general formula CnH2n
1. Homologous series of alkenes
|
WITH n H 2 n alkene |
Names, suffix EH, ILENE |
|
C2H4 |
this en, this Ilen |
|
C3H6 |
propene |
|
C4H8 |
butene |
|
C5H10 |
penten |
|
C6H12 |
hexene |
Homologues:
WITHH 2 = CH 2 ethene
WITHH 2 = CH- CH 3 propene
WITHH 2 =CH-CH 2 -CH 3butene-1
WITHH 2 =CH-CH 2 -CH 2 -CH 3 penten-1
2. Physical properties
Ethylene (ethene) – colorless gas with a very faint sweetish odor, slightly lighter than air, slightly soluble in water.
C 2 – C 4 (gases)
C 5 – C 17 (liquids)
C 18 – (solid)
· Alkenes are insoluble in water, soluble in organic solvents (gasoline, benzene, etc.)
Lighter than water
With increasing Mr, the melting and boiling points increase
3. The simplest alkene is ethylene - C2H4
Structural and electronic formula ethylene have the form:
In the ethylene molecule one undergoes hybridization s- and two p-orbitals of C atoms ( sp 2 -hybridization).
Thus, each C atom has three hybrid orbitals and one non-hybrid p-orbitals. Two of the hybrid orbitals of the C atoms mutually overlap and form between the C atoms
σ - bond. The remaining four hybrid orbitals of the C atoms overlap in the same plane with four s-orbitals of H atoms and also form four σ - bonds. Two non-hybrid p-orbitals of C atoms mutually overlap in a plane that is located perpendicular to the σ-bond plane, i.e. one is formed P- connection.


By it's nature P- connection is sharply different from σ - connection; P- the bond is less strong due to the overlap of electron clouds outside the plane of the molecule. Under the influence of reagents P- the connection is easily broken.
The ethylene molecule is symmetrical; the nuclei of all atoms are located in the same plane and bond angles are close to 120°; the distance between the centers of C atoms is 0.134 nm.
If atoms are connected by a double bond, then their rotation is impossible without electron clouds P- the connection was not opened.
4. Isomerism of alkenes
Along with structural isomerism of the carbon skeleton Alkenes are characterized, firstly, by other types of structural isomerism - multiple bond position isomerism And interclass isomerism.
Secondly, in the series of alkenes there is spatial isomerism , associated with different positions of substituents relative to double bond, around which intramolecular rotation is impossible.
Structural isomerism of alkenes
1. Isomerism of the carbon skeleton (starting from C 4 H 8):
2. Isomerism of the position of the double bond (starting from C 4 H 8):
3. Interclass isomerism with cycloalkanes, starting with C 3 H 6:
Spatial isomerism of alkenes
Rotation of atoms around a double bond is impossible without breaking it. This is due to the structural features of the p-bond (the p-electron cloud is concentrated above and below the plane of the molecule). Due to the rigid attachment of atoms rotational isomerism does not appear regarding the double bond. But it becomes possible cis-trance-isomerism.
Alkenes, which have different substituents on each of the two carbon atoms at the double bond, can exist in the form of two spatial isomers, differing in the location of the substituents relative to the plane of the p-bond. So, in the butene-2 molecule CH 3 –CH=CH–CH 3 CH 3 groups can be located either on one side of the double bond in cis-isomer, or different sides V trance-isomer.

ATTENTION!
cis-trans- Isomerism does not appear if at least one of the C atoms at the double bond has 2 identical substituents.
For example,
butene-1 CH 2 = CH – CH 2 – CH 3 doesn't have cis- And trance-isomers, because The 1st C atom is bonded to two identical H atoms.
Isomers cis- And trance- differ not only physically
 ,
,
but also chemical properties, because bringing parts of a molecule closer or further away from each other in space promotes or hinders chemical interaction.
Sometimes cis-trans-isomerism is not quite accurately called geometric isomerism . The inaccuracy is that All spatial isomers differ in their geometry, and not only cis- And trance-.
5. Nomenclature
Alkenes simple structure often called by replacing the suffix -ane in alkanes with -ylene: ethane - ethylene, propane - propylene, etc.
By systematic nomenclature The names of ethylene hydrocarbons are made by replacing the suffix -ane in the corresponding alkanes with the suffix -ene (alkane - alkene, ethane - ethene, propane - propene, etc.). The choice of the main chain and the naming order are the same as for alkanes. However, the chain must necessarily include a double bond. The numbering of the chain begins from the end to which this connection is located closest. For example:

Unsaturated (alkene) radicals are called by trivial names or by systematic nomenclature:
(H 2 C=CH-)vinyl or ethenyl
(H 2 C=CH-CH 2) allyl
General formula alkenes: CnH2n(n 2)
The first representatives of the homologous series of alkenes:

The formulas of alkenes can be made from the corresponding formulas of alkanes ( saturated hydrocarbons). The names of alkenes are formed by replacing the suffix -ane of the corresponding alkane with -ene or –ylene: butane - butylene, pentane - pentene, etc. The number of the carbon atom with a double bond is indicated by an Arabic numeral after the name.
The carbon atoms involved in the formation of the double bond are in a state of sp-hybridization. Three -bonds formed by hybrid orbitals and are located in the same plane at an angle of 120° to each other. An additional -bond is formed by lateral overlap of non-hybrid p-orbitals:

The length of the C=C double bond (0.133 nm) is less than the length single bond(0.154 nm). The energy of a double bond is less than twice the energy of a single bond because the energy of the -bond is less than the energy of the -bond.
Alkene isomers
All alkenes except ethylene have isomers. Alkenes are characterized by isomerism of the carbon skeleton, isomerism of the position of the double bond, interclass and spatial isomerism.
The interclass isomer of propene (C 3 H 6) is cyclopropane. Starting with butene (C 4 H 8), isomerism appears at the position of the double bond (butene-1 and butene-2), isomerism of the carbon skeleton (methylpropene or isobutylene), as well as spatial isomerism(cis-butene-2 and trans-butene-2). In cis isomers, the substituents are located on one side, and in trans isomers, they are located on opposite sides of the double bond.
Chemical properties And chemical activity alkenes are determined by the presence of a double bond in their molecules. The most common reactions for alkenes are electrophilic addition: hydrohalogenation, hydration, halogenation, hydrogenation, polymerization.
Qualitative reaction to a double bond - discoloration bromine water:
Examples of solving problems on the topic “formula of alkenes”
EXAMPLE 1
| Exercise | How many isomers capable of decolorizing bromine water does a substance with the composition C 3 H 5 Cl have? Write structural formulas these isomers |
| Solution | C 3 H 5 Cl is a monochlor derivative of the hydrocarbon C 3 H 6 . This formula corresponds to either propene, a hydrocarbon with one double bond, or cyclopropane (a cyclic hydrocarbon). This substance discolors bromine water, which means it contains a double bond. Three carbon atoms can only form this structure: since isomerism of the carbon skeleton and the position of the double bond is impossible with such a number of carbon atoms. Structural isomerism in a given molecule is possible only due to a change in the position of the chlorine atom relative to the double bond: For 1-chloropropene, cis-trans isomerism is possible: |
| Answer | The problem conditions are satisfied by 4 isomers |
EXAMPLE 2
| Exercise | A mixture of isomeric hydrocarbons (gases with a hydrogen density of 21) with a volume of 11.2 l (n.s.) reacted with bromine water. The result was 40.4 g of the corresponding dibromo derivative. What structure do these hydrocarbons have? Determine their volumetric content in the mixture (in%). |
| Solution | The general formula of hydrocarbons is C x H y. Let's calculate the molar mass of hydrocarbons: Therefore, the formula of hydrocarbons is C 3 H 6. Only two substances have this formula - propene and cyclopropane. Only propene reacts with bromine water: Let's calculate the amount of dibromo derivative substance: According to the reaction equation: n(propene) mol The total amount of hydrocarbons in the mixture is equal to: |
4. Chemical properties of alkenes
The energy of a double carbon-carbon bond in ethylene (146 kcal/mol) turns out to be significantly lower than twice the energy of a single C-C bond in ethane (2 88 = 176 kcal/mol). -C-C bond in ethylene stronger connections, therefore, reactions of alkenes accompanied by the cleavage of an -bond with the formation of two new simple -bonds are a thermodynamically favorable process. For example, in the gas phase, according to calculated data, all the reactions below are exothermic with a significant negative enthalpy, regardless of their actual mechanism.
From the point of view of the theory of molecular orbitals, it can also be concluded that the -bond is more reactive than the -bond. Let's consider the molecular orbitals of ethylene (Fig. 2).

Indeed, the bonding orbital of ethylene has more high energy, than the bonding -orbital, and vice versa, the antibonding *-orbital of ethylene lies below the antibonding *-orbital of the C=C bond. Under normal conditions, the *- and *-orbitals of ethylene are vacant. Consequently, the boundary orbitals of ethylene and other alkenes that determine them reactivity there will be -orbitals.
4.1. Catalytic hydrogenation of alkenes
Despite the fact that the hydrogenation of ethylene and other alkenes to alkanes is accompanied by the release of heat, this reaction occurs at a noticeable rate only in the presence of certain catalysts. The catalyst, by definition, does not affect the thermal effect of the reaction, and its role is reduced to reducing the activation energy. It is necessary to distinguish between heterogeneous and homogeneous catalytic hydrogenation of alkenes. In heterogeneous hydrogenation, finely ground metal catalysts are used - platinum, palladium, ruthenium, rhodium, osmium and nickel, or pure form, or deposited on inert carriers - BaSO 4, CaCO 3, activated carbon, Al 2 O 3, etc. All of them are insoluble in organic environments and act as heterogeneous catalysts. The most active among them are ruthenium and rhodium, but platinum and nickel are most widespread. Platinum is usually used in the form of black dioxide PtO 2, commonly known as Adams catalyst. Platinum dioxide is obtained by fusing chloroplatinic acid H 2 PtCl 6 . 6H 2 O or ammonium hexachloroplatinate (NH 4) 2 PtCl 6 with sodium nitrate. The hydrogenation of alkenes with an Adams catalyst is usually carried out at normal pressure and a temperature of 20-50 0 C in alcohol, acetic acid, ethyl acetate. When hydrogen is passed through, platinum dioxide is reduced directly in the reaction vessel to platinum black, which catalyzes hydrogenation. Other more active platinum group metals are used on inert supports, for example, Pd/C or Pd/BaSO 4, Ru/Al 2 O 3; Rh/C, etc. Palladium supported on coal catalyzes the hydrogenation of alkenes to alkanes in an alcohol solution at 0-20 0 C and normal pressure. Nickel is usually used in the form of so-called "Raney nickel". To obtain this catalyst, a nickel-aluminum alloy is treated with hot aqueous alkali to remove almost all aluminum and then with water until a neutral reaction. The catalyst has a porous structure and is therefore also called a skeletal nickel catalyst. Typical conditions for the hydrogenation of alkenes over Raney nickel require the use of a pressure of the order of 5-10 atm and a temperature of 50-100 0 C, i.e. this catalyst is much less active than platinum group metals, but it is cheaper. Below are some typical examples of heterogeneous catalytic hydrogenation of acyclic and cyclic alkenes:

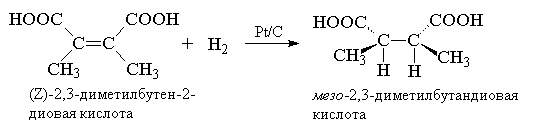
Since both hydrogen atoms are added to the carbon atoms of the double bond from the surface of the catalyst metal, the addition usually occurs on one side of the double bond. This type of connection is called syn- accession. In cases where two reagent fragments are added together various sides multiple bond (double or triple) occurs anti- accession. Terms syn- And anti- are equivalent in meaning to the terms cis- And trance-. To avoid confusion and misunderstanding the terms syn- And anti- refer to the type of connection, and the terms cis- And trance- to the structure of the substrate.
The double bond in alkenes is hydrogenated at a higher rate compared to many other functional groups (C=O, COOR, CN, etc.) and therefore hydrogenation of the C=C double bond is often a selective process if the hydrogenation is carried out in mild conditions(0-20 0 C and at atmospheric pressure). Below are some typical examples:


The benzene ring is not reduced under these conditions.
Big and important important achievement in catalytic hydrogenation is the discovery of soluble metal complexes that catalyze hydrogenation in a homogeneous solution. Heterogeneous hydrogenation on the surface of metal catalysts has a number of significant disadvantages, such as isomerization of alkenes and cleavage of single carbon-carbon bonds (hydrogenolysis). Homogeneous hydrogenation does not have these disadvantages. Behind last years A large group of homogeneous hydrogenation catalysts - transition metal complexes containing various ligands - has been obtained. The best catalysts for homogeneous hydrogenation are complexes of rhodium (I) and ruthenium (III) chlorides with triphenylphosphine - tris(triphenylphosphine)rhodium chloride (Ph 3 P) 3 RhCl (Wilkinson's catalyst) and tris(triphenylphosphine) ruthenium hydrochloride (Ph 3 P) 3 RuHCl. The most accessible rhodium complex is obtained by reacting rhodium(III) chloride with triphenylphosphine. Wilkinson's rhodium complex is used to hydrogenate the double bond in normal conditions.

An important advantage of homogeneous catalysts is the ability to selectively reduce a mono- or disubstituted double bond in the presence of a tri- and tetra-substituted double bond due to the large differences in their hydrogenation rates.

In the case of homogeneous catalysts, hydrogen addition also occurs as syn- accession. So recovery cis-butene-2 with deuterium under these conditions leads to meso-2,3-dideuterobutane.

4.2. Reduction of a double bond using diimide
The reduction of alkenes to the corresponding alkanes can be successfully accomplished using diimide NH=NH.
Diimide is obtained by two main methods: the oxidation of hydrazine with hydrogen peroxide in the presence of Cu 2+ ions or the reaction of hydrazine with Ni-Raney (hydrazine dehydrogenation). If an alkene is present in the reaction mixture, its double bond is hydrogenated by the highly unstable diimide. A distinctive feature of this method is the strict syn-stereospecificity of the restoration process. It is believed that this reaction proceeds through a cyclic activated complex with a strict orientation of both reacting molecules in space.

4.3. Electrophilic addition reactions at the double bond of alkenes
The boundary HOMO and LUMO orbitals of alkenes are the occupied and empty * orbitals. Consequently, the -orbital will participate in reactions with electrophiles (E +), and the *-orbital of the C=C bond will participate in reactions with nucleophiles (Nu -) (see Fig. 3). In most cases, simple alkenes react easily with electrophiles, but react with nucleophiles with great difficulty. This is explained by the fact that usually the LUMO of most electrophiles is close in energy to the energy of the -HOMO of alkenes, while the HOMO of most nucleophiles lies significantly below the *-LUMO.

Simple alkenes react only with very strong nucleophilic agents (carbanions) under harsh conditions, however, the introduction of electron-withdrawing groups into alkenes, for example, NO 2, COR, etc., leads to a decrease in the * level, due to which the alkene acquires the ability to react with nucleophiles of average strength (ammonia, RO - , Nє C - , enolate anion, etc.).
As a result of the interaction of the electrophilic agent E + with an alkene, a carbocation is formed, which is highly reactive. The carbocation is further stabilized by the rapid addition of the nucleophilic agent Nu - :


Since the slow stage is the addition of an electrophile, the process of addition of any polar agent E + Nu - should be considered precisely as an electrophilic addition to the multiple bond of an alkene. A large number of reactions of this type are known, where the role of the electrophilic agent is played by halogens, hydrogen halides, water, divalent mercury salts and other polar reagents. Electrophilic addition to a double bond in the classification of mechanisms organic reactions has the symbol Ad E ( Addition Electrophilic) and, depending on the number of reacting molecules, is designated as Ad E 2 (bimolecular reaction) or Ad E 3 (trimolecular reaction).
4.3.a. Addition of halogens
Alkenes react with bromine and chlorine to form addition products at the double bond of one halogen molecule with a yield close to quantitative. Fluorine is too active and causes the destruction of alkenes. The addition of iodine to alkenes in most cases is reversible reaction, the equilibrium of which is shifted towards the original reagents.


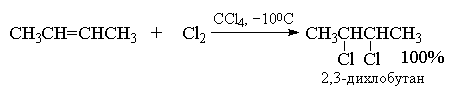
The rapid decolorization of a solution of bromine in CCl4 serves as one of the simplest tests for unsaturation, since alkenes, alkynes, and dienes react quickly with bromine.
The addition of bromine and chlorine to alkenes occurs by an ionic rather than a radical mechanism. This conclusion follows from the fact that the rate of halogen addition does not depend on irradiation, the presence of oxygen and other reagents that initiate or inhibit radical processes. Based on a large number of experimental data, a mechanism was proposed for this reaction, including several sequential stages. At the first stage, polarization of the halogen molecule occurs under the action of bonding electrons. The halogen atom, which acquires a certain fractional positive charge, forms an unstable intermediate with the electrons of the -bond, called an -complex or a charge transfer complex. It should be noted that in the -complex the halogen does not form a directed bond with any specific carbon atom; In this complex, the donor-acceptor interaction of an electron pair - bond as a donor and a halogen as an acceptor is simply realized.

Next, the -complex transforms into a cyclic bromonium ion. During the formation of this cyclic cation, heterolytic cleavage occurs Br-Br bonds and empty R-the sp 2 orbital of the hybridized carbon atom overlaps with R-orbital of the “lone pair” of electrons of the halogen atom, forming a cyclic bromonium ion.

In the last, third stage, the bromine anion, as a nucleophilic agent, attacks one of the carbon atoms of the bromonium ion. Nucleophilic attack by the bromide ion leads to the opening of the three-membered ring and the formation of a vicinal dibromide ( vic-near). This step can formally be considered as a nucleophilic substitution of SN 2 at the carbon atom, where the leaving group is Br+.
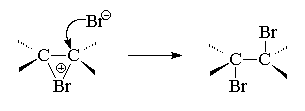
The addition of halogens to the double bond of alkenes is one of the formally simple model reactions, using the example of which one can consider the influence of the main factors that allow one to draw reasoned conclusions about the detailed mechanism of the process. To make informed conclusions about the mechanism of any reaction, you should have data on: 1) reaction kinetics; 2) stereochemistry (stereochemical result of the reaction); 3) the presence or absence of an associated, competing process; 4) the influence of substituents in the original substrate on the reaction rate; 5) use of labeled substrates and (or) reagents; 6) the possibility of rearrangements during the reaction; 7) the effect of the solvent on the reaction rate.
Let us consider these factors using the example of the halogenation of alkenes. Kinetic data make it possible to establish the order of the reaction for each component and, on this basis, draw a conclusion about the overall molecularity of the reaction, i.e., the number of reacting molecules.
For the bromination of alkenes, the reaction rate is typically described by the following equation:
v = k`[alkene] + k``[alkene] 2,
which in in rare cases simplifies to
v = k`[alkene].
Based on the kinetic data, it can be concluded that one or two bromine molecules are involved in the rate-determining step. The second order in bromine means that it is not the bromide ion Br - that reacts with the bromonium ion, but the tribromide ion formed by the interaction of bromine and bromide ion:
![]()
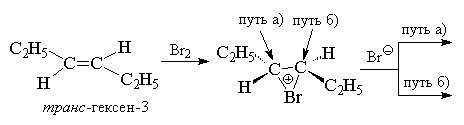
This balance is shifted to the right. Kinetic data do not allow any other conclusions about the structure transition state and the nature of the electrophilic particle in the reaction of halogen addition at the double bond. The most valuable information about the mechanism of this reaction is provided by data on the stereochemistry of the addition. The addition of a halogen to a double bond is a stereospecific process (a process in which only one of the possible stereoisomers is formed; in a stereoselective process, the preferential formation of one stereomer is observed) anti-additions for alkenes and cycloalkenes, in which the double bond is not conjugated with benzene ring. For cis- And trance-isomers of butene-2, pentene-2, hexene-3, cyclohexene, cyclopentene and other alkenes, the addition of bromine occurs exclusively as anti- accession. In this case, in the case of cyclohexene, only trance-1,2-dibromocyclohexane (mixture of enantiomers).

The trans arrangement of bromine atoms in 1,2-dibromocyclohexane can be depicted in a simplified manner relative to the average plane of the cyclohexane ring (without taking into account conformations):
When bromine combines with cyclohexene, it initially forms trance-1,2-dibromocyclohexane in a,a-conformation, which then immediately transforms into an energetically more favorable her-conformation. Anti-the addition of halogens to a double bond allows us to reject the mechanism of one-step synchronous addition of one halogen molecule to a double bond, which can only occur as syn- accession. Anti-addition of a halogen is also inconsistent with the formation of an open carbocation RCH + -CH 2 Hal as an intermediate. In an open carbocation, free rotation around the C-C bond is possible, which should lead to the attack of the Br anion - to the formation of a mixture of products as anti- and so syn- accessions. Stereospecific anti-the addition of halogens was main reason creating the concept of bromonium or chloronium ions as discrete intermediate species. This concept perfectly satisfies the rule anti-addition, since nucleophilic attack of the halide ion is possible with anti-sides at either of the two carbon atoms of the halide ion via the S N 2 mechanism.

In the case of unsymmetrically substituted alkenes, this should result in two enantiomers trio-form upon addition of bromine to cis-isomer or enantiomer erythro-forms upon halogenation trance-isomer. This is actually observed when bromine is added to, for example, cis- And trance-isomers of pentene-2.
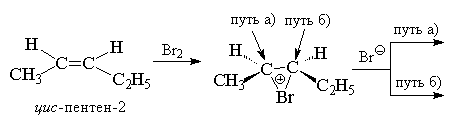



In the case of bromination of symmetrical alkenes, for example, cis- or trance-hexene-3 should be formed or a racemate ( D,L-form), or meso-form of the final dibromide, which is what is actually observed.



There is independent, direct evidence of the existence of halogenium ions in a non-nucleophilic, indifferent environment at low temperature. Using NMR spectroscopy, the formation of bromonium ions was recorded during the ionization of 3-bromo-2-methyl-2-fluorobutane under the action of very strong acid Lewis antimony pentafluoride in a solution of liquid sulfur dioxide at -80 0 C.

This cation is quite stable at -80 0 C in a non-nucleophilic environment, but is instantly destroyed by the action of any nucleophilic agents or upon heating.
Cyclic bromonium ions can sometimes be isolated in pure form if steric obstacles prevent their opening under the action of nucleophiles:

It is clear that the possibility of the existence of bromonium ions, which are quite stable under special conditions, cannot serve as direct evidence of their formation in the reaction of bromine addition to the double bond of an alkene in alcohol, acetic acid and other electron-donating solvents. Such data should be considered only as independent confirmation of the fundamental possibility of the formation of halogenium ions in the process of electrophilic addition at the double bond.
The concept of the halide ion allows us to provide a rational explanation for the reversibility of the addition of iodine to the double bond. The halogenium cation has three electrophilic centers accessible to nucleophilic attack by the halide anion: two carbon atoms and a halogen atom. In the case of chloronium ions, the Cl - anion appears to preferentially or even exclusively attack the carbon centers of the cation. For the bromonium cation, both directions of opening of the halogenium ion are equally probable, both due to the attack of the bromide ion on both carbon atoms and on the bromine atom. Nucleophilic attack on the bromine atom of the bromonium ion leads to the starting reagents bromine and alkene:

The iodonium ion is revealed predominantly as a result of the attack of the iodide ion on the iodine atom, and therefore the equilibrium between the starting reagents and the iodonium ion is shifted to the left.

In addition, the final addition product, vicinal diiodide, can be subject to nucleophilic attack at the iodine atom by the triiodide anion present in the solution, which also leads to the formation of the initial reagents alkene and iodine. In other words, under the conditions of the addition reaction, the resulting vicinal diiodide is deiodinated under the action of the triiodide anion. Vicinal dichlorides and dibromides do not dehalogenate under the conditions of the addition of chlorine or bromine, respectively, to alkenes.
Anti-addition of chlorine or bromine is characteristic of alkenes, in which the double bond is not conjugated with the -electrons of the benzene ring. For styrene, stilbene and their derivatives along with anti- accession takes place and syn-addition of a halogen, which can even become dominant in a polar environment.

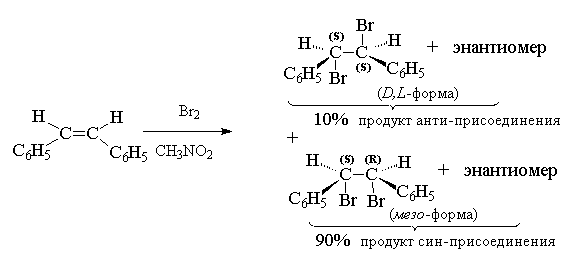
In cases where the addition of a halogen to a double bond is carried out in a nucleophilic solvent environment, the solvent effectively competes with the halide ion in opening the three-membered ring of the halogenium ion:

The formation of addition products with the participation of a solvent or some other “external” nucleophilic agent is called a conjugate addition reaction. When bromine and styrene react in methanol, two products are formed: vicinal dibromide and bromine ester, the ratio of which depends on the concentration of bromine in methanol
In a highly dilute solution, the conjugate addition product dominates, while in a concentrated solution, on the contrary, vicinal dibromide predominates. In an aqueous solution, halohydrin (an alcohol containing a halogen at the -carbon atom) - the product of conjugate addition - always predominates.

her-conformer trance-2-chlorocyclohexanol is further stabilized by an O-H hydrogen bond . . . Cl. In the case of unsymmetrical alkenes, in conjugate addition reactions, the halogen always adds to the carbon atom containing the largest number of hydrogen atoms, and the nucleophilic agent to the carbon with the least number of hydrogen atoms. An isomeric product with a different arrangement of joining groups is not formed. This means that the cyclic halogenonium ion formed as an intermediate must have an asymmetric structure with two bonds C 1 -Hal and C 2 -Hal differing in energy and strength and more positive charge on the internal carbon atom C2, which can be expressed graphically in two ways:

Therefore, the C2 carbon atom of the halogenium ion is subject to nucleophilic attack by the solvent, despite the fact that it is more substituted and sterically less accessible.


One of the best preparative methods for the synthesis of bromohydrins is the hydroxybromination of alkenes using N-bromosuccinimide ( N.B.S.) in a binary mixture of dimethyl sulfoxide ( DMSO) and water.

This reaction can be carried out in water or without DMSO, however, the yields of bromohydrins in this case are somewhat lower.



The formation of conjugate addition products in the halogenation reaction of alkenes also allows us to reject the synchronous mechanism of addition of one halogen molecule. Conjugate addition to the double bond is in good agreement with a two-step mechanism involving the halogenium cation as an intermediate.
For the reaction of electrophilic addition to a double bond, one should expect an increase in the reaction rate in the presence of electron-donating alkyl substituents and a decrease in the presence of electron-withdrawing substituents at the double bond. Indeed, the rate of addition of chlorine and bromine to the double bond increases sharply when moving from ethylene to its methyl-substituted derivatives. For example, the rate of addition of bromine to tetramethylethylene is 10 5 times higher than the rate of its addition to 1-butene. This enormous acceleration clearly indicates the high polarity of the transition state and the high degree of charge separation in the transition state and is consistent with the eletrophilic mechanism of addition.
In some cases, the addition of chlorine to alkenes containing electron-donating substituents is accompanied by the abstraction of a proton from the intermediate compound instead of the addition of a chloride ion. The abstraction of a proton results in the formation of a chlorine-substituted alkene, which can formally be considered as a direct substitution with double bond migration. However, experiments with isotope labeling indicate more complex nature transformations taking place here. When isobutylene is chlorinated at 0 0 C, 2-methyl-3-chloropropene (metallyl chloride) is formed instead of the expected dichloride, the product of addition at the double bond.

Formally, it seems as if there is a substitution, not an accession. The study of this reaction using isobutylene labeled at position 1 with the 14 C isotope showed that direct replacement of hydrogen with chlorine does not occur, since in the resulting metallyl chloride the label is located in the 14 CH 2 Cl group. This result can be explained by the following sequence of transformations:

In some cases, 1,2-migration of the alkyl group may also occur

In CCl 4 (non-polar solvent) this reaction gives almost 100% dichloride B- product of ordinary addition at a double bond (without rearrangement).
Skeletal rearrangements of this type are most typical for processes involving open carbocations as intermediate particles. It is possible that the addition of chlorine in these cases occurs not through the chloronium ion, but through a cationic particle close to the open carbocation. At the same time, it should be noted that skeletal rearrangements are a rather rare phenomenon in the processes of addition of halogens and mixed halogens at the double bond: they are more often observed during the addition of chlorine and much less frequently during the addition of bromine. The probability of such rearrangements increases when moving from non-polar solvents (CCl 4) to polar ones (nitromethane, acetonitrile).
Summarizing the presented data on stereochemistry, conjugate addition, the influence of substituents in the alkene, as well as rearrangements in the addition reactions of halogens at the double bond, it should be noted that they are in good agreement with the mechanism of electrophilic addition involving the cyclic halogenium ion. Data on the addition of mixed halogens to alkenes, for which the stages of addition are determined by the polarity of the bond of two halogen atoms, can be interpreted in the same way.
DEFINITION
Alkenes are called unsaturated hydrocarbons, whose molecules contain one double bond. The structure of the alkene molecule using ethylene as an example is shown in Fig. 1.
Rice. 1. The structure of the ethylene molecule.
By physical properties alkenes differ little from alkanes with the same number of carbon atoms in the molecule. Lower homologs C 2 - C 4 at normal conditions- gases; C 5 - C 17 - liquids; higher homologs - solids. Alkenes are insoluble in water. Highly soluble in organic solvents.
Preparation of alkenes
In industry, alkenes are obtained during oil refining: cracking and dehydrogenation of alkanes. Laboratory methods We divided the production of alkenes into two groups:
- Elimination reactions
– dehydration of alcohols
CH 3 -CH 2 -OH → CH 2 =CH 2 + H 2 O (H 2 SO 4 (conc), t 0 = 170).
— dehydrohalogenation of monohaloalkanes
CH 3 -CH(Br)-CH 2 -CH 3 + NaOH alcohol → CH 3 -CH=CH-CH 3 + NaBr + H 2 O (t 0).
— dehalogenation of dihaloalkanes
CH 3 -CH(Cl)-CH(Cl)-CH 2 -CH 3 + Zn(Mg) → CH 3 -CH=CH-CH 2 -CH 3 + ZnCl 2 (MgCl 2).
- Incomplete hydrogenation of alkynes
CH≡CH + H 2 →CH 2 =CH 2 (Pd, t 0).
Chemical properties of alkenes
Alkenes are highly reactive organic compounds. This is explained by their structure. The chemistry of alkenes is the chemistry of double bonds. Typical reactions for alkenes - electrophilic addition reactions.
Chemical transformations of alkenes proceed with splitting:
1) π - S-S connections(addition, polymerization and oxidation)
- hydrogenation
CH 3 -CH=CH 2 + H 2 → CH 3 -CH 2 -CH 2 (kat = Pt).
- halogenation
CH 3 -CH 2 -CH=CH 2 + Br 2 → CH 3 -CH 2 -CH(Br)-CH 2 Br.
— hydrohalogenation (proceeds according to Markovnikov’s rule: a hydrogen atom attaches preferentially to a more hydrogenated carbon atom)
CH 3 -CH=CH 2 + H-Cl → CH 3 -CH(Cl)-CH 3 .
- hydration
CH 2 =CH 2 + H-OH → CH 3 -CH 2 -OH (H + , t 0).
- polymerization
nCH 2 =CH 2 → -[-CH 2 -CH 2 -]- n (kat, t 0).
- oxidation
CH 2 =CH 2 + 2KMnO 4 + 2KOH → HO-CH 2 -CH 2 -OH + 2K 2 MnO 4;
2CH 2 =CH 2 + O 2 → 2C 2 OH 4 (epoxide) (kat = Ag,t 0);
2CH 2 =CH 2 + O 2 → 2CH 3 -C(O)H (kat = PdCl 2, CuCl).
2) σ- and π-C-C bonds
CH 3 -CH=CH-CH 2 -CH 3 + 4[O] → CH 3 COOH + CH 3 CH 2 COOH (KMnO 4, H +, t 0).
3) bonds C sp 3 -H (in the allylic position)
CH 2 =CH 2 + Cl 2 → CH 2 =CH-Cl + HCl (t 0 =400).
4) Breaking all ties
C 2 H 4 + 2O 2 → 2CO 2 + 2H 2 O;
C n H 2n + 3n/2 O 2 → nCO 2 + nH 2 O.
Applications of alkenes
Alkenes have found use in various industries National economy. Let's look at the example of individual representatives.
Ethylene is widely used in industrial organic synthesis to produce a variety of organic compounds, such as halogen derivatives, alcohols (ethanol, ethylene glycol), acetaldehyde, acetic acid etc. B large quantities ethylene is used to produce polymers.
Propylene is used as a raw material for the production of some alcohols (for example, 2-propanol, glycerin), acetone, etc. Polypropylene is produced by polymerization of propylene.
Examples of problem solving
EXAMPLE 1
| Exercise | During hydrolysis aqueous solution sodium hydroxide NaOH dichloride, obtained by adding 6.72 l of chlorine to ethylene hydrocarbon, formed 22.8 g dihydric alcohol. What is the formula of the alkene if it is known that the reactions proceed in quantitative yields (without losses)? |
| Solution | Let us write the equation for alkene chlorination in general view, as well as the reaction to produce dihydric alcohol: C n H 2 n + Cl 2 = C n H 2 n Cl 2 (1); C n H 2 n Cl 2 + 2NaOH = C n H 2 n (OH) 2 + 2HCl (2). Let's calculate the amount of chlorine: n(Cl 2) = V(Cl 2) / V m; n(Cl 2) = 6.72 / 22.4 = 0.3 mol, therefore, ethylene dichloride will also be 0.3 mol (equation 1), dihydric alcohol should also be 0.3 mol, and according to the conditions of the problem this is 22.8 g. This means molar mass it will be equal to: M(C n H 2 n (OH) 2) = m(C n H 2 n (OH) 2) / n(C n H 2 n (OH) 2); M(C n H 2 n (OH) 2) = 22.8 / 0.3 = 76 g/mol. Let's find the molar mass of the alkene: M(C n H 2 n) = 76 - (2×17) = 42 g/mol, which corresponds to the formula C 3 H 6 . |
| Answer | Alkene formulaC 3 H 6 |
EXAMPLE 2
| Exercise | How many grams will be required to brominate 16.8 g of an alkene, if it is known that during the catalytic hydrogenation of the same amount of alkene, 6.72 liters of hydrogen were added? What is the composition and possible structure of the original hydrocarbon? |
| Solution | Let us write in general form the equations for the bromination and hydrogenation of an alkene: C n H 2 n + Br 2 = C n H 2 n Br 2 (1); C n H 2 n + H 2 = C n H 2 n +2 (2). Let's calculate the amount of hydrogen substance: n(H 2) = V(H 2) / V m; n(H 2) = 6.72 / 22.4 = 0.3 mol, therefore, the alkene will also be 0.3 mol (equation 2), and according to the conditions of the problem it is 16.8 g. This means its molar mass will be equal to: M(C n H 2n) = m(C n H 2n) / n(C n H 2n); M(C n H 2 n) = 16.8 / 0.3 = 56 g/mol, which corresponds to the formula C 4 H 8 . According to equation (1) n(C n H 2 n) : n(Br 2) = 1:1, i.e. n(Br 2) = n(C n H 2 n) = 0.3 mol. Let's find the mass of bromine: m(Br 2) = n(Br 2) × M(Br 2); M(Br 2) = 2×Ar(Br) = 2×80 = 160 g/mol; m(MnO 2) = 0.3 × 160 = 48 g. Let's create the structural formulas of the isomers: butene-1 (1), butene-2 (2), 2-methylpropene (3), cyclobutane (4). CH 2 =CH-CH 2 -CH 3 (1); CH 3 -CH=CH-CH 3 (2); CH 2 =C(CH 3)-CH 3 (3); |
| Answer | The mass of bromine is 48 g |
The physical properties of alkenes are similar to those of alkanes, although they all have slightly more low temperatures melting and boiling than the corresponding alkanes. For example, pentane has a boiling point of 36 °C, and pentene-1 - 30 °C. Under normal conditions, alkenes C 2 - C 4 are gases. C 5 – C 15 are liquids, starting from C 16 are solids. Alkenes are insoluble in water but highly soluble in organic solvents.
Alkenes are rare in nature. Since alkenes are valuable raw materials for industrial organic synthesis, many ways to obtain them have been developed.
1. The main industrial source of alkenes is the cracking of alkanes that are part of oil:
3. B laboratory conditions Alkenes are obtained by elimination reactions, in which two atoms or two groups of atoms are eliminated from neighboring carbon atoms, and an additional p-bond is formed. Such reactions include the following.
1) Dehydration of alcohols occurs when they are heated with water-removing agents, for example with sulfuric acid at temperatures above 150 ° C:
When H 2 O is eliminated from alcohols, HBr and HCl from alkyl halides, the hydrogen atom is preferentially eliminated from that of the neighboring carbon atoms that is bonded to the smallest number hydrogen atoms (from the least hydrogenated carbon atom). This pattern is called Zaitsev's rule.
3) Dehalogenation occurs when dihalides that have halogen atoms at adjacent carbon atoms are heated with active metals:
CH 2 Br -CHBr -CH 3 + Mg → CH 2 =CH-CH 3 + Mg Br 2.
The chemical properties of alkenes are determined by the presence of a double bond in their molecules. Electron density p-bonds are quite mobile and easily react with electrophilic particles. Therefore, many reactions of alkenes proceed according to the mechanism electrophilic addition, designated by the symbol A E (from English, addition electrophilic). Electrophilic addition reactions are ionic processes that occur in several stages.
In the first stage, an electrophilic particle (most often this is an H + proton) interacts with the p-electrons of the double bond and forms a p-complex, which is then converted into a carbocation by forming a covalent s-bond between the electrophilic particle and one of the carbon atoms:
alkene p-complex carbocation
In the second stage, the carbocation reacts with the X - anion, forming a second s-bond due to the electron pair of the anion:

In electrophilic addition reactions, a hydrogen ion attaches to the carbon atom at the double bond that has the largest number of carbon atoms. negative charge. The charge distribution is determined by the shift in p-electron density under the influence of substituents:  .
.
Electron-donating substituents exhibiting the +I effect shift the p-electron density to a more hydrogenated carbon atom and create a partial negative charge on it. This explains Markovnikov's rule: when adding polar molecules like HX (X = Hal, OH, CN, etc.) to unsymmetrical alkenes, hydrogen preferentially attaches to the more hydrogenated carbon atom at the double bond.
Let's consider specific examples addition reactions.
1) Hydrohalogenation. When alkenes interact with hydrogen halides (HCl, HBr), alkyl halides are formed:
CH 3 -CH = CH 2 + HBr ® CH 3 -CHBr-CH 3 .
The reaction products are determined by Markovnikov's rule.
It should be emphasized, however, that in the presence of any organic peroxide polar HX molecules react with alkenes not according to Markovnikov’s rule:
| R-O-O-R | ||
| CH 3 -CH = CH 2 + HBr | CH 3 -CH 2 -CH 2 Br |
This is due to the fact that the presence of peroxide causes radical rather than ion mechanism reactions.
2) Hydration. When alkenes react with water in the presence of mineral acids (sulfuric, phosphoric), alcohols are formed. Mineral acids act as catalysts and are sources of protons. The addition of water also follows Markovnikov’s rule:
CH 3 -CH = CH 2 + HON ® CH 3 -CH (OH) -CH 3 .
3) Halogenation. Alkenes discolor bromine water:
CH 2 = CH 2 + Br 2 ® B-CH 2 -CH 2 Br.
This reaction is qualitative for a double bond.
4) Hydrogenation. The addition of hydrogen occurs under the action of metal catalysts:
where R = H, CH 3, Cl, C 6 H 5, etc. The CH 2 =CHR molecule is called a monomer, the resulting compound is called a polymer, the number n is the degree of polymerization.
Polymerization of various alkene derivatives gives valuable industrial products: polyethylene, polypropylene, polyvinyl chloride and others.
In addition to addition, alkenes also undergo oxidation reactions. During the mild oxidation of alkenes with an aqueous solution of potassium permanganate (Wagner reaction), dihydric alcohols are formed:
ZSN 2 =CH 2 + 2KMn O 4 + 4H 2 O ® ZNOSN 2 -CH 2 OH + 2MnO 2 ↓ + 2KOH.
As a result of this reaction, the purple solution of potassium permanganate quickly becomes discolored and a brown precipitate of manganese (IV) oxide precipitates. This reaction, like the decolorization reaction of bromine water, is qualitative for a double bond. During the severe oxidation of alkenes with a boiling solution of potassium permanganate in an acidic environment, complete cleavage of the double bond occurs with the formation of ketones, carboxylic acids or CO 2, for example:
| [ABOUT] | ||
| CH 3 -CH=CH-CH 3 | 2CH 3 -COOH |
Based on the oxidation products, the position of the double bond in the original alkene can be determined.
Like all other hydrocarbons, alkenes burn and, with plenty of air, form carbon dioxide and water:
C n H 2 n + Zn /2O 2 ® n CO 2 + n H 2 O.
When air is limited, combustion of alkenes can lead to the formation of carbon monoxide and water:
C n H 2n + nO 2 ® nCO + nH 2 O .
If you mix an alkene with oxygen and pass this mixture over a silver catalyst heated to 200°C, an alkene oxide (epoxyalkane) is formed, for example:

At any temperature, alkenes are oxidized by ozone (ozone is a stronger oxidizing agent than oxygen). If ozone gas is passed through a solution of an alkene in methane tetrachloride at temperatures below room temperature, an addition reaction occurs and the corresponding ozonides (cyclic peroxides) are formed. Ozonides are very unstable and can explode easily. Therefore, they are usually not isolated, but immediately after production they are decomposed with water - this produces carbonyl compounds(aldehydes or ketones), the structure of which indicates the structure of the alkene that was subjected to ozonation.
Lower alkenes are important starting materials for industrial organic synthesis. From ethylene it is obtained ethanol, polyethylene, polystyrene. Propene is used for the synthesis of polypropylene, phenol, acetone, and glycerin.